铁死亡与脓毒性休克
铁死亡 脓毒性休克
## 铁死亡在脓毒性休克中的病理生理机制与治疗潜力
铁死亡是一种铁依赖性的、由脂质过氧化驱动的调节性细胞死亡形式。其核心机制涉及**谷胱甘肽(GSH)耗竭**和**谷胱甘肽过氧化物酶4(GPX4)活性丧失**,导致脂质过氧化物(如丙二醛,MDA)积累,最终引发细胞死亡[1]。形态学上,其特征为**线粒体皱缩、嵴减少或消失、双层膜密度增加**,而无凋亡的典型特征[1]。
### 铁死亡与脓毒症/脓毒性休克的关联证据
现有证据表明,铁死亡参与了脓毒症及其并发症的病理过程,可能成为新的治疗靶点。
**1. 脓毒症相关性脑病(SAE)**
* **动物模型证据**:在盲肠结扎穿孔(CLP)诱导的脓毒症小鼠模型中,观察到脑内铁死亡标志物增加,包括**GPX4失活、转铁蛋白上调、线粒体收缩和MDA水平升高**[2]。
* **机制假说**:铁死亡可能通过促进谷氨酸释放,参与谷氨酸介导的兴奋性毒性神经元损伤[2]。
* **干预效果**:使用铁死亡抑制剂**铁抑素-1(Fer-1)** 可下调脑内铁死亡,减轻谷氨酸兴奋性毒性,保护突触和神经元完整性,并改善脓毒症小鼠的行为学表现[2]。这提示抑制铁死亡可能改善SAE预后。
**2. 急性呼吸窘迫综合征(ARDS)**
* **动物模型证据**:在脂多糖(LPS)诱导的ARDS大鼠模型中,铁调素(hepcidin)被证实可以减轻肺部炎症和损伤[2]。
* **作用机制**:铁调素通过**上调铁蛋白重链表达、降低铁摄入、增加铁储备**,发挥类似于Fer-1的作用,抑制铁死亡的发生[2]。这表明靶向铁代谢可能通过抑制铁死亡来改善脓毒症相关的肺损伤。
**3. 作为潜在的治疗策略**
一篇综述指出,靶向调节性细胞死亡(包括铁死亡)机制在脓毒症治疗中具有广阔前景[2]。这为超越传统抗感染和支持治疗,从细胞死亡通路层面进行干预提供了理论依据。
### 临床意义与未来方向
1. **生物标志物与分型**:铁死亡相关标志物(如脂质过氧化物、GPX4活性)可能有助于识别特定的脓毒症内毒素反应表型或预测器官损伤(如SA-AKI)[2]。早期识别不同脓毒症表型有助于个体化治疗策略的研究。
2. **治疗靶点**:铁螯合剂(如去铁胺)、抗氧化剂(如Fer-1)以及调节铁代谢的分子(如铁调素)是潜在的干预手段[1][2]。然而,**所有相关证据均来自临床前动物研究**,其安全性和有效性在人体中尚未得到证实。
3. **研究现状**:目前关于铁死亡在脓毒性休克中的作用研究仍处于**基础探索阶段**。证据主要来源于小鼠和大鼠模型,缺乏人体临床试验数据。其具体机制、在脓毒症多器官功能障碍中的普遍性、以及最佳干预时机和靶点均有待深入阐明。
### 结论
铁死亡是连接脓毒症免疫炎症反应与细胞损伤的重要程序性死亡途径之一,尤其与脓毒症相关性脑病和急性肺损伤的病理过程密切相关。临床前研究提示,抑制铁死亡可能具有器官保护作用。然而,这目前仍是一个**高度活跃的基础研究领域**,距离转化为成熟的临床治疗方案尚有很长的路要走。未来的研究方向包括在更大型的动物模型中验证疗效、探索可靠的人类生物标志物,并最终设计严谨的临床试验进行评估。
---
*本分析基于提供的文献资料生成,旨在总结当前基础研究进展。所有提及的干预措施均处于临床前研究阶段,不构成临床治疗建议。具体的诊疗决策需结合最新临床指南和患者个体情况。*
血管内皮损伤
## 血管内皮损伤:脓毒性休克的核心病理生理机制
血管内皮损伤是脓毒性休克发生发展的核心环节,它连接了感染、炎症、凝血紊乱和器官功能障碍。
### 内皮损伤的病理生理机制
**1. 直接损伤因素**
* **病原体相关分子模式(PAMPs)**:如细菌脂多糖(LPS),通过模式识别受体直接激活内皮细胞。
* **损伤相关分子模式(DAMPs)**:由坏死细胞释放,进一步放大炎症反应[8]。
* **中性粒细胞胞外诱捕网(NETs)**:在脓毒症中大量形成,可直接损伤内皮细胞并激活凝血系统[8]。
**2. 内皮糖萼降解**
内皮糖萼是覆盖于血管内皮腔面的多糖-蛋白复合物层,是维持血管屏障功能和抗凝状态的关键结构。
* **标志物**:其降解产物**血浆硫酸乙酰肝素(HS)和Syndecan-1**是内皮损伤的敏感指标[1][8]。
* **临床意义**:在CLOVERS试验的事后分析中,基线内皮糖萼降解程度(通过HS和Syndecan-1测量)是脓毒症患者**90天死亡率的强预测因子**(具体HR值未在提供上下文中给出)[1]。糖萼降解导致血管通透性增加、白细胞黏附增强和促凝状态。
**3. 内皮细胞功能表型转换**
正常内皮细胞具有抗凝、抗血小板聚集和维持血管张力的功能。在脓毒症时,其表型转为促炎、促凝。
| 正常功能 (抗凝/抗炎) | 损伤后功能 (促凝/促炎) |
| :--- | :--- |
| **屏障作用**:分隔血液成分与内皮下基质[6] | **通透性增加**:导致组织水肿和低血容量[13][14] |
| **合成PGI₂、NO**:抑制血小板黏附[6] | **释放vWF**:介导血小板黏附[6] |
| **合成血栓调节蛋白(TM)**:激活蛋白C抗凝系统[6][7] | **释放组织因子(TF)**:启动外源性凝血途径[6][8] |
| **合成组织型纤溶酶原激活物(t-PA)**:促进纤溶[6] | **分泌纤溶酶原激活物抑制物(PAI-1)**:抑制纤溶[6] |
| **表达硫酸乙酰肝素**:增强抗凝血酶Ⅲ作用[7] | **糖萼脱落**:失去上述保护功能[1] |
### 内皮损伤的临床后果
**1. 血流动力学紊乱(脓毒性休克)**
* **血管舒张与低血压**:内皮损伤导致NO等舒血管物质过量产生,同时对血管收缩剂反应性降低(血管麻痹)[9][13]。
* **毛细血管渗漏**:屏障功能丧失导致大量液体外渗,加重有效循环血容量不足[13][14]。
* **微循环障碍**:内皮细胞肿胀、微血栓形成等导致功能性毛细血管密度减少,组织氧利用障碍[14]。
**2. 凝血功能紊乱(脓毒症性凝血病,SIC)**
内皮损伤是SIC的始动因素。根据《脓毒症性凝血病诊疗中国专家共识(2024版)》,其过程可分为两个阶段[4][5]:
**以下流程图展示了脓毒症性凝血病(SIC)从高凝到低凝的动态演变过程:**
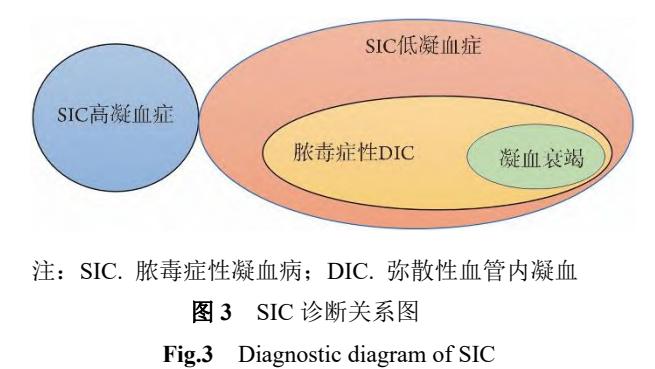
*Caption: 该流程图详细阐述了脓毒症诱发凝血功能障碍(SIC)从病原体入侵到多器官损伤及出血的病理生理演变过程。*
* **SIC高凝期**:内皮损伤释放TF,同时TM、TFPI等抗凝物质下调,导致凝血酶大量生成(TAT升高),血液呈高凝状态。临床可表现为隐匿性器官功能损害或诱发血栓事件[4]。
* **SIC低凝期/脓毒症性DIC**:持续高凝导致凝血因子和血小板大量消耗,进入消耗性低凝状态,可合并纤溶抑制(t-PAIC升高)或纤溶亢进(PIC升高),临床出现多器官衰竭和出血倾向[4][5]。
**3. 多器官功能障碍综合征(MODS)**
广泛的内皮损伤被认为是MODS的主要机制[13][14]。微循环障碍和微血栓形成导致组织灌注不足和缺血缺氧,进而引起器官功能衰竭。
### 内皮损伤的评估与监测
**1. 分子标志物**
* **血栓调节蛋白(TM)**:内皮细胞表面受体,损伤时脱落入血,是内皮损伤的敏感指标。需注意肾功能不全时TM也会升高[3][8]。
* **组织型纤溶酶原激活物-纤溶酶原激活物抑制物-1复合物(t-PAIC)**:反映内皮严重损伤和纤溶抑制,常提示休克相关内皮病[4][8]。
* **血管性血友病因子(vWF)**:内皮细胞储存和释放,水平升高提示内皮激活或损伤。
* **内皮糖萼成分**:如Syndecan-1、硫酸乙酰肝素(HS)[1][8]。
**2. 功能评估**
* **血管通透性评估**:如血管外肺水指数(EVLWI)。
* **微循环监测**:如舌下微循环成像(正交偏振光谱成像),可直观评估微血管血流[14]。
### 治疗启示与管理策略
**1. 血流动力学管理**
* **早期使用血管活性药物**:去甲肾上腺素是脓毒性休克的一线升压药,旨在维持平均动脉压(MAP)≥65 mmHg,以保障器官灌注压[9]。
* **限制性液体策略**:CLOVERS试验的事后分析发现,**宽松与限制性液体复苏策略对患者的内皮糖萼降解程度和死亡率影响无显著差异**[1]。这提示过度液体复苏可能无益,甚至可能加重内皮水肿和器官损伤。
**2. 针对内皮保护的潜在策略**
* **抗炎治疗**:如糖皮质激素,可能通过减轻炎症反应间接保护内皮。
* **抗凝治疗**:治疗SIC/DIC,如使用抗凝血酶、血栓调节蛋白制剂或活化蛋白C(需权衡出血风险)。
* **血管紧张素Ⅱ**:用于难治性血管麻痹性休克,可能有助于恢复血管张力。
* **保护糖萼的药物**:如硫酸乙酰肝素类似物、硒(GPX4辅因子)等尚在研究中。
**3. 支持治疗**
* **器官支持**:如机械通气、肾脏替代治疗等,为内皮修复争取时间。
* **控制感染源**:根本措施。
### 总结
血管内皮损伤是脓毒性休克“血管麻痹、毛细血管渗漏、凝血紊乱、器官衰竭”这一系列病理改变的共同通路。监测内皮损伤标志物(如TM、t-PAIC)有助于评估病情严重程度和预后[3][8]。当前治疗核心仍是早期抗感染、精细化的血流动力学支持(包括合理的液体管理和血管活性药使用)以及器官功能支持。针对内皮和糖萼的特异性保护疗法是未来重要的研究方向。
---
*本分析基于提供的文献资料生成,旨在总结病理生理机制与现有证据。具体的临床决策需结合最新指南、患者个体情况及动态监测指标。*
脓毒性休克血管内皮损伤 铁死亡 生脉注射液
## 脓毒性休克中血管内皮损伤、铁死亡与生脉注射液的关联分析
本分析将整合脓毒性休克中血管内皮损伤与铁死亡的病理生理联系,并基于现有证据评估生脉注射液在此背景下的潜在作用机制与临床应用。
### 核心病理生理联系:内皮损伤与铁死亡
脓毒性休克中,**血管内皮损伤**与**铁死亡**并非孤立事件,而是存在相互促进的恶性循环。
1. **内皮损伤是铁死亡的触发因素之一**:
* **炎症与氧化应激**:活化的内皮细胞释放大量促炎细胞因子(如TNF-α)和活性氧(ROS),这些物质可直接攻击细胞膜脂质,诱发脂质过氧化,这是铁死亡的核心环节[2][14]。
* **微循环障碍与缺血缺氧**:内皮损伤导致的毛细血管渗漏和微血栓形成,造成组织灌注不足和缺血缺氧。缺氧环境可诱导**缺氧诱导因子-1α(HIF-1α)** 上调,进而增加细胞对铁的摄取和储存,为铁死亡创造条件[1]。
2. **铁死亡加剧内皮损伤**:
* **内皮细胞死亡**:血管内皮细胞本身可发生铁死亡。铁死亡导致的脂质过氧化产物(如MDA)和细胞内容物释放,会进一步损伤邻近的内皮细胞,扩大损伤范围。
* **屏障功能丧失**:铁死亡破坏内皮细胞的完整性,直接导致血管通透性增加,加重组织水肿和休克[1][14]。
* **促凝状态**:死亡的内皮细胞暴露内皮下胶原,释放促凝物质,加剧脓毒症性凝血病(SIC)[4][6]。
**结论**:在脓毒性休克中,感染引发的全身炎症首先导致内皮损伤,进而通过氧化应激和微循环障碍等途径诱发或加重包括内皮细胞在内的多种实质细胞的铁死亡;反之,铁死亡又进一步破坏内皮屏障和功能,形成正反馈循环,共同推动多器官功能障碍的发展[1][2][14]。
### 生脉注射液的药理作用与潜在靶点
生脉注射液由红参、麦冬、五味子组成,具有益气养阴、复脉固脱之功效。根据提供的证据,其作用机制可能从多个环节干预上述病理过程。
**1. 保护血管内皮**
* **实验证据**:生脉注射液中的**人参皂苷**被证实是其保护血管内皮细胞抵抗缺氧/复氧损伤的主要活性部位[5]。
* **潜在机制**:可能通过减少活性氧生成、降低乳酸脱氢酶(LDH)和丙二醛(MDA)水平,减轻氧化应激对内皮细胞的损伤[5]。
**2. 抗休克与改善微循环**
* **临床研究证据**:
* 针对感染性休克患者,生脉注射液可**改善氧代谢、调节血清细胞因子、降低炎症因子水平**,并调节内皮素-1和一氧化氮的表达,从而提高临床总有效率[5]。
* 对于脓毒性休克(气阴亏虚型)患者,注射用益气复脉(冻干)(生脉类注射剂)联合常规西医治疗的疗效优于单纯西医治疗[5]。
* 在感染性休克治疗中,联合使用注射用益气复脉(冻干)与重酒石酸去甲肾上腺素,可于较短时间内降低患者的血乳酸水平[5]。
* **动物实验机制**:生脉注射液的抗休克作用与其抑制脂多糖(LPS)刺激导致的诱导型一氧化氮合酶(iNOS)过度表达、调节免疫功能、改善微循环有关[5]。
**3. 潜在的抗铁死亡作用**
* **间接证据与理论关联**:虽然现有文献未直接研究生脉注射液对铁死亡的影响,但其已知的药理作用可能间接对抗铁死亡的关键环节:
* **抗氧化**:其降低MDA、提高超氧化物歧化酶(SOD)活力的作用[5],可直接对抗铁死亡的核心驱动因素——脂质过氧化。
* **改善能量代谢**:生脉注射液可改善心肌细胞缺氧耐受性和线粒体功能[5]。铁死亡与线粒体功能障碍和ATP产生减少密切相关,改善能量代谢可能有助于细胞抵抗铁死亡。
* **“补气固摄”与减少渗出**:研究发现其成分可改善LPS引起的微血管渗出,抑制血浆白蛋白和水的漏出[5]。这有助于维持血管内容量,改善微循环灌注,从而缓解组织缺血缺氧,减少铁死亡的诱因。
### 临床证据总结与应用考量
| 证据类别 | 具体发现 | 证据等级与来源 |
| :--- | :--- | :--- |
| **内皮保护** | 人参皂苷是保护血管内皮免受缺氧/复氧损伤的活性成分。 | 实验研究[5] |
| **抗休克疗效** | 可改善感染性休克患者氧代谢、炎症指标及临床有效率。 | 小样本临床研究[5] |
| **联合治疗优势** | 联合常规西医治疗对气阴亏虚型脓毒性休克患者疗效更优。 | 小样本RCT[5] |
| **改善血流动力学** | 联合去甲肾上腺素可更快降低感染性休克患者血乳酸。 | 临床研究提及[5] |
| **作用机制** | 与抑制iNOS、调节免疫、改善微循环、抗氧化有关。 | 实验与综述[5] |
**临床定位与使用建议**:
1. **辅助治疗地位**:所有证据均支持生脉注射液作为**脓毒性休克/感染性休克的辅助治疗药物**,应在规范的西医抗感染、液体复苏、血管活性药等基础治疗之上使用[3][5]。
2. **中医辨证应用**:其适应症为**气阴两亏、脉虚欲脱**的证候[7]。对于“热毒炽盛”等实证,需谨慎或禁用[7]。
3. **用法用量参考**:
* 生脉注射液静脉滴注:一次20-60ml,以5%葡萄糖注射液250-500ml稀释后使用[7]。
* 撤药顺序:当血流动力学稳定需撤药时,建议**血管活性药物应先于中药注射剂撤药**[3]。
4. **证据局限性**:现有临床研究多为小样本试验,缺乏大规模、多中心的随机对照试验(RCT)提供高级别证据。
### 结论与展望
脓毒性休克中的血管内皮损伤与铁死亡构成了相互加剧的病理轴。生脉注射液通过其**抗氧化、保护内皮、改善微循环和调节免疫**的多重药理作用,可能同时作用于这一轴线的多个环节,从而在辅助治疗中发挥效益。其临床疗效已有初步研究支持,但证据强度有待提升。未来研究可深入探索生脉注射液是否直接抑制内皮细胞的铁死亡,并开展更严谨的临床试验,以明确其在脓毒性休克综合治疗中的最佳定位和价值。
---
*本分析基于提供的文献资料生成。生脉注射液的使用需严格遵循中医辨证原则和药品说明书,并在全面西医支持治疗的基础上进行。临床决策应结合患者具体情况和最新指南。*
脓毒性休克中血管内皮细胞铁死亡的具体分子机制与信号通路
## 脓毒性休克中血管内皮细胞铁死亡的具体分子机制与信号通路
基于现有证据,脓毒性休克中血管内皮细胞(Endothelial Cells, ECs)发生铁死亡是一个由**炎症、氧化应激和铁代谢紊乱**共同驱动的复杂过程。其核心机制涉及**抗氧化防御系统崩溃**和**脂质过氧化级联反应**的激活。
### 核心分子机制图解
**以下示意图整合了脓毒性休克中诱导血管内皮细胞铁死亡的关键信号通路:**
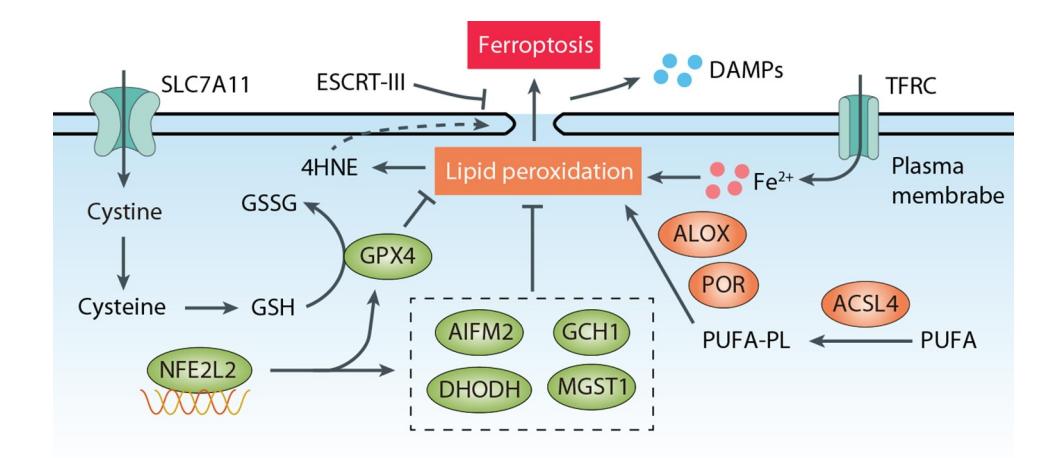
*Caption: 该图展示了铁死亡的核心调控网络,包括系统Xc-/GPX4抗氧化轴、铁代谢以及脂质过氧化驱动通路,为理解内皮细胞铁死亡提供了分子框架。*
### 一、 抗氧化防御系统失活(铁死亡的核心抑制通路被破坏)
这是启动铁死亡的关键前提。内皮细胞的抗氧化能力在脓毒症中被严重削弱。
1. **系统Xc-/GSH/GPX4轴抑制**:
* **SLC7A11(系统Xc-关键组分)下调**:在脓毒症炎症环境下,促炎因子(如TNF-α, IL-1β)可抑制**SLC7A11**的表达或活性[1]。这导致**胱氨酸(Cystine)** 摄取减少,进而使合成**谷胱甘肽(GSH)** 的原料**半胱氨酸(Cysteine)** 匮乏。
* **GSH耗竭**:GSH是细胞内最重要的抗氧化剂之一,也是**GPX4**的必需辅因子。GSH耗竭直接导致GPX4活性丧失[1]。
* **GPX4降解或失活**:
* **直接氧化失活**:脓毒症产生的大量活性氧(ROS)可直接氧化GPX4的活性位点硒代半胱氨酸,使其失活。
* **自噬依赖性降解**:如图2所示,GPX4可通过**伴侣介导的自噬(CMA)** 或**选择性自噬(由受体TAX1BP1介导)** 途径被降解[8]。脓毒症时自噬活动紊乱,可能促进这一过程。
2. **GPX4非依赖性防御系统受损**:
* **FSP1/CoQ10系统**:位于质膜的**FSP1(AIFM2)** 能还原泛醌(CoQ10)为泛醇(CoQ10H2),后者是一种脂溶性抗氧化剂,可捕获脂质过氧自由基。脓毒症可能干扰此系统。
* **GCH1/BH4系统**:**GCH1**是合成四氢生物蝶呤(BH4)的限速酶,BH4及其代谢产物具有抗氧化活性。该系统在内皮细胞中尤为重要,其功能障碍与内皮氧化损伤密切相关。
### 二、 脂质过氧化驱动系统激活(铁死亡的执行通路)
当抗氧化防御崩溃,富含多不饱和脂肪酸(PUFA)的磷脂(PUFA-PLs)被过氧化,导致膜损伤。
1. **脂质底物供应(ACSL4/LPCAT3)**:
* **ACSL4**被上调或激活,它催化长链PUFAs(如花生四烯酸AA、肾上腺酸AdA)与辅酶A结合,生成PUFA-CoAs[1]。
* **LPCAT3**等酶将这些PUFA-CoAs酯化到膜磷脂(特别是磷脂酰乙醇胺PE)上,形成易于过氧化的底物PUFA-PLs。
2. **脂质过氧化启动(LOXs/ POR / ROS)**:
* **脂氧合酶(ALOXs, 特别是ALOX15)**:某些ALOX亚型(如ALOX15)可直接氧化膜上的PUFA-PLs,生成脂质氢过氧化物(PLOOH)[1]。ALOX15在内皮细胞中有表达,且其表达可能受炎症信号调控。
* **细胞色素P450氧化还原酶(POR)**:POR可通过产生ROS间接引发脂质过氧化[1]。
* **芬顿反应(Fenton Reaction)**:这是**铁依赖性的核心环节**。脓毒症时,细胞内**不稳定铁池(LIP)** 增加,Fe²⁺与H₂O₂反应生成高毒性的**羟基自由基(·OH)**,直接攻击脂质。
### 三、 铁代谢紊乱(铁死亡的“燃料”供应)
细胞内铁超载是铁死亡的标志和必要条件。
1. **铁摄取增加**:
* **转铁蛋白受体1(TFRC)** 表达上调,增加转铁蛋白结合铁的摄取[1]。
* 脓毒症低氧环境诱导**HIF-1α**,其下游靶基因包括促进铁摄取的基因。
2. **铁储存释放(铁蛋白自噬,Ferritinophagy)**:
* 这是脓毒症背景下尤为重要的铁来源。**NCOA4**作为选择性自噬受体,识别并结合铁蛋白(Ferritin),将其靶向至自噬溶酶体降解,从而释放出Fe²⁺[8]。
* **以下示意图详细展示了NCOA4介导的铁蛋白自噬如何驱动铁死亡:**

*Caption: 该图阐明了NCOA4介导的铁蛋白自噬(Ferritinophagy)通路,展示了从铁储存释放到引发脂质过氧化的完整链条。*
* 脓毒症相关信号(如氧化应激、炎症)可能调控NCOA4的表达或活性,加剧铁蛋白自噬。
3. **铁输出受阻**:
* 膜铁转运蛋白(FPN1)的表达可能下调或功能受损,导致细胞内铁排出减少。
### 四、 脓毒症特异性信号通路的交叉对话
脓毒症的全身炎症反应与上述核心通路深度交织:
* **TLR4/NF-κB通路**:LPS通过TLR4激活NF-κB,产生大量促炎因子(TNF-α, IL-6),这些因子可抑制SLC7A11转录,同时诱导ROS产生。
* **炎症小体激活**:NLRP3炎症小体激活后产生的IL-1β等,可进一步加剧氧化应激和内皮功能障碍。
* **HMGB1**:作为重要的DAMP,HMGB1可通过其受体(如RAGE)促进内皮细胞炎症和氧化损伤,可能参与铁死亡的调控。
### 总结:脓毒性休克内皮细胞铁死亡的级联反应
1. **触发**:病原体(LPS)及炎症介质通过TLR4/NF-κB等通路激活内皮细胞,导致ROS爆发和炎症因子释放。
2. **准备**:炎症信号抑制SLC7A11,导致GSH合成减少;同时,通过铁蛋白自噬(NCOA4介导)和TFRC上调导致细胞内Fe²⁺超载。
3. **失衡**:GPX4因GSH缺乏和/或自噬降解而失活,抗氧化防御崩溃。同时,ACSL4/ALOXs等脂质过氧化驱动系统被激活。
4. **执行**:Fe²⁺通过芬顿反应催化产生·OH,与ALOXs等共同氧化膜上的PUFA-PLs,产生大量脂质过氧化物(如MDA, 4-HNE)。
5. **死亡**:脂质过氧化物积累导致质膜和细胞器膜(尤其是线粒体膜)完整性破坏,离子梯度丧失,最终细胞破裂死亡,释放DAMPs,进一步放大全身炎症和内皮损伤。
**治疗启示**:针对该机制,潜在的治疗靶点包括:**补充GSH前体(如N-乙酰半胱氨酸)、使用铁螯合剂(去铁胺)、GPX4激动剂或模拟物、FSP1激动剂、抑制ACSL4或ALOX15、以及调控NCOA4介导的铁蛋白自噬**。然而,所有这些策略目前主要处于临床前研究阶段。
---
*本分析基于提供的铁死亡与自噬相关文献生成,阐述了理论上的分子机制通路。这些机制在人体脓毒性休克中的确切作用和权重仍需进一步实验验证。临床转化前需进行严格的药理学和安全性评估。*
与线粒体自噬 代谢重编程
## 脓毒性休克中血管内皮细胞铁死亡与线粒体自噬、代谢重编程的交互网络
在脓毒性休克中,血管内皮细胞的铁死亡并非孤立事件,而是与**线粒体自噬(Mitophagy)**和**代谢重编程**构成一个相互影响、动态演变的复杂网络。三者共同决定了内皮细胞的命运(存活或死亡)和器官功能障碍的进程。
### 一、 线粒体自噬与铁死亡的“双刃剑”关系
线粒体自噬是选择性清除受损线粒体的过程,在内皮细胞应对脓毒症应激时扮演着矛盾的角色。
**1. 保护性线粒体自噬(抑制铁死亡)**
* **机制**:及时清除功能失调、产生过量ROS的线粒体,可以**从源头上减少ROS和脂质过氧化物的产生**,从而保护细胞免于铁死亡[3][8]。
* **关键通路**:**PINK1-PRKN(Parkin)通路**是经典的线粒体自噬启动通路。
* **正常情况**:PINK1被持续导入健康线粒体并降解。
* **线粒体损伤(如脓毒症)**:线粒体膜电位(ΔΨm)下降,PINK1在线粒体外膜稳定积累,募集并激活胞质中的E3泛素连接酶PRKN[4]。
* **执行自噬**:PRKN泛素化线粒体外膜蛋白,被自噬受体(如OPTN, NDP52)识别,进而与LC3结合,将受损线粒体包裹进自噬体进行降解[4]。
**以下实验证据直观展示了PINK1-PRKN通路在启动线粒体自噬中的核心作用:**
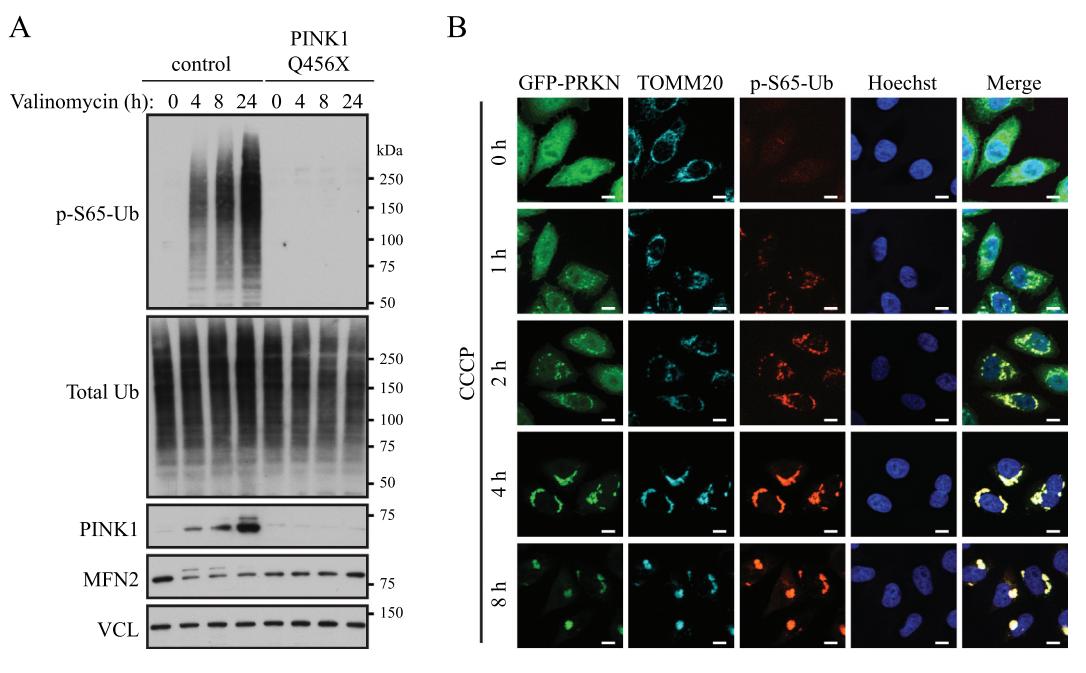
*Caption: Western blot与免疫荧光实验证实,线粒体去极化(Valinomycin/CCCP处理)导致PINK1稳定、Ubiquitin磷酸化(p-S65-Ub)及PRKN向线粒体募集,此过程被PINK1 Q456X突变完全阻断。*
* **与铁死亡的关联**:功能正常的线粒体自噬通过清除ROS源头,间接维持了GPX4等抗氧化系统的功能,是细胞抵抗铁死亡的重要防御机制[3]。
**2. 过度或失调的线粒体自噬(促进铁死亡)**
* **机制**:当线粒体损伤过于广泛或自噬过程失控时,过度自噬可能导致**线粒体数量严重不足**。
* **后果**:
* **能量危机**:线粒体是ATP主要来源,其大量丢失导致细胞能量(ATP)耗竭。
* **代谢失衡**:影响包括脂质合成与氧化在内的多种代谢途径。
* **促进铁死亡**:ATP严重耗竭本身可诱发细胞死亡。更重要的是,线粒体是**铁硫簇合成和血红素代谢**的主要场所。线粒体功能崩溃会扰乱细胞铁代谢,可能导致**不稳定铁池(LIP)** 异常升高,为芬顿反应提供充足底物,从而**强力驱动铁死亡**[1][8]。
* **证据支持**:研究提示,在某些情况下,线粒体自噬相关蛋白(如PINK1、FUNDC1)的激活反而与铁死亡敏感性增加相关[3]。这可能反映了“适度清除有益,过度清除有害”的双相效应。
### 二、 代谢重编程:铁死亡的“引擎”与“燃料库”
脓毒症中,内皮细胞经历深刻的代谢重编程,从**以氧化磷酸化(OXPHOS)为主**转向**糖酵解增强(Warburg效应)**,这直接影响了铁死亡的易感性。
**1. 糖酵解增强与抗氧化能力削弱**
* **PPP途径分流减少**:糖酵解亢进可能减少葡萄糖流向**磷酸戊糖途径(PPP)**。PPP是生成**NADPH**的主要途径之一。
* **关键影响**:NADPH是维持**GSH还原状态(GSH再生)** 和**FSP1/CoQ10系统**功能的关键辅因子[1]。NADPH供应不足,直接导致**GPX4和FSP1两大铁死亡防御系统失活**,使细胞对脂质过氧化极度敏感。
**2. 脂质代谢重编程:直接提供铁死亡底物**
* **脂肪酸合成与摄取增加**:脓毒症炎症信号(如TLR4激活)可上调内皮细胞中**脂肪酸合成酶(FASN)** 的表达,并促进**CD36**等脂肪酸转运蛋白介导的脂质摄取[1]。
* **后果**:细胞内**游离多不饱和脂肪酸(PUFAs)** 池扩大。在**ACSL4**的催化下,这些PUFAs被活化为PUFA-CoAs,并进一步酯化到膜磷脂上,为铁死亡提供了丰富的脂质过氧化底物[1]。
**3. 铁代谢与三羧酸循环(TCA)紊乱**
* **线粒体功能障碍**:脓毒症导致线粒体TCA循环酶活性受损,代谢中间产物积累。
* **意外后果**:某些TCA循环中间产物的积累(如延胡索酸、琥珀酸)可抑制**α-酮戊二酸依赖性双加氧酶**,从而稳定**HIF-1α**并激活**NCOA4**介导的**铁蛋白自噬**,导致铁释放增加[1][8]。
### 三、 三者交互作用的整合模型
脓毒性休克中,内皮细胞的命运由以下动态网络决定:
```
脓毒症应激(LPS/炎症因子)
↓
[代谢重编程]
├─ 糖酵解增强 → NADPH生成减少 → GPX4/FSP1系统抑制
├─ 脂质合成/摄取增加 → PUFA-PLs底物丰富
└─ TCA循环紊乱 → 激活铁蛋白自噬 → 细胞内铁超载
↓
[线粒体损伤]
├─ ROS大量产生 → 脂质过氧化启动
└─ 膜电位下降 → 激活PINK1-PRKN通路
↓
[线粒体自噬激活] → 双刃剑效应
├─ 适度自噬:清除ROS源头 → **抑制铁死亡**
└─ 过度自噬:铁代谢紊乱/能量耗竭 → **促进铁死亡**
↓
[铁死亡执行]
抗氧化防御(GPX4等)崩溃 + 脂质过氧化驱动(Fe²⁺/LOXs)增强
↓
内皮细胞铁死亡 → 血管屏障破坏、促凝、炎症放大
```
**总结与治疗启示**:
1. **网络视角**:在脓毒性休克中,不应孤立看待铁死亡。**代谢重编程**为其准备了“燃料”(PUFA, Fe²⁺)和削弱了“防火墙”(抗氧化系统);**线粒体自噬**的平衡则决定了是清除“火源”(ROS)还是耗尽“能源”(ATP)并加剧“燃料泄漏”(铁释放)。
2. **动态性与阶段性**:在休克早期,适度的代谢适应和线粒体自噬可能是保护性的;随着病理过程失控,三者可能形成正反馈循环,加速内皮崩溃。
3. **潜在干预靶点**:
* **调控代谢**:改善内皮细胞糖代谢流向(如促进PPP),或调节脂质代谢(如抑制ACSL4或CD36)。
* **精准调控自噬**:旨在**增强保护性线粒体自噬的精确性**(如通过Urolithin A等线粒体自噬诱导剂),同时**避免过度自噬和铁蛋白自噬**。这是一个精细的平衡策略。
* **联合阻断**:针对铁死亡核心通路(如铁螯合、GPX4保护)与调控代谢/自噬相结合,可能是更有效的内皮保护策略。
---
*本分析基于铁死亡、自噬及代谢相关文献的理论整合。该交互网络在人体脓毒症中的具体调控细节及作为治疗靶点的安全窗,仍需大量基础与临床研究验证。任何干预都需考虑脓毒症复杂的全身病理生理背景。*
OCT4 SOX2与卵巢子宫内膜样癌 铁死亡
## OCT4、SOX2与卵巢子宫内膜样癌及铁死亡的关联分析
基于提供的文献,目前**没有直接证据**表明**OCT4**和**SOX2**与**卵巢子宫内膜样癌(Ovarian Endometrioid Carcinoma)** 的**铁死亡(Ferroptosis)** 过程存在明确关联。现有信息指向两个独立的领域:OCT4/SOX2是**卵巢生殖细胞肿瘤**的诊断标志物,而卵巢子宫内膜样癌的预后和治疗有其自身特点。
### 一、 OCT4与SOX2的明确临床意义
根据《病理学(第10版)》,**OCT4**和**SOX2**是**卵巢胚胎性癌(Embryonal Carcinoma)** 的关键诊断标志物。
1. **肿瘤类型**:胚胎性癌是一种**高度恶性的原始生殖细胞肿瘤**,多见于儿童和年轻女性,与起源于体腔上皮的**卵巢子宫内膜样癌**在组织起源、发病年龄、生物学行为上完全不同[1]。
2. **诊断价值**:在病理诊断中,肿瘤细胞表达**OCT4**和**CD30**或**SOX2**阳性,有助于确诊胚胎性癌[1]。这是其作为**生殖细胞来源**和**未分化状态**的标志。
3. **与子宫内膜样癌的关系**:在常规诊断中,OCT4和SOX2**不用于**卵巢子宫内膜样癌的诊断或分型。子宫内膜样癌通常表达**雌激素受体(ER)**、**孕激素受体(PR)** 和**PAX8**等标志物。
### 二、 卵巢子宫内膜样癌的临床特征与治疗
根据《Blaustein‘s Pathology of the Female Genital Tract》,卵巢子宫内膜样癌具有以下特点:
1. **预后**:总体预后优于卵巢高级别浆液性癌,主要原因是**早期(I期)病例比例高**。I期患者的5年无病生存率可达95%[2]。
2. **治疗**:主要治疗模式与其他上皮性卵巢癌相同,即肿瘤细胞减灭术联合以铂类为基础的化疗[2]。
3. **特殊治疗选择**:对于复发或无法耐受化疗的患者,**激素治疗**(如孕激素、抗雌激素药物如他莫昔芬)可能作为一种选择,尤其当肿瘤组织表达类固醇激素受体时,但疗效有限[2]。
### 三、 关于“铁死亡”的关联性分析
1. **在卵巢癌中的研究现状**:铁死亡作为一种铁依赖性的程序性细胞死亡方式,在多种癌症(包括卵巢癌)中被研究其治疗潜力。诱导铁死亡可能成为克服化疗耐药的新策略。
2. **与OCT4/SOX2的理论关联(研究前沿推测)**:
* **干细胞性与治疗抵抗**:在**其他癌种**(如胶质母细胞瘤、肺癌)的研究中,OCT4和SOX2作为维持肿瘤干细胞(CSCs)干性和自我更新的核心转录因子,与化疗耐药和复发相关。
* **潜在机制假说**:肿瘤干细胞通常处于代谢相对静止状态,抗氧化能力较强(如高表达GSH、GPX4),这可能使其对诱导铁死亡不敏感。因此,靶向OCT4/SOX2通路**可能**通过调控下游代谢和抗氧化相关基因,影响肿瘤细胞对铁死亡的敏感性。
* **重要说明**:**这是一个基于其他肿瘤研究的外推假说**。在**卵巢子宫内膜样癌**中,OCT4和SOX2是否高表达、是否与干细胞特性相关、以及是否调控铁死亡通路,**均未见诸于提供的文献,也非当前该癌种的常规认知**。
### 四、 结论与临床意义
1. **诊断与分类**:**OCT4和SOX2是卵巢胚胎性癌(生殖细胞肿瘤)的诊断标志物,而非卵巢子宫内膜样癌的标志物**。两者在临床病理诊断中用于区分完全不同的肿瘤类型。
2. **治疗**:卵巢子宫内膜样癌的治疗遵循上皮性卵巢癌原则,对激素治疗可能有部分反应。目前的标准治疗或挽救性治疗方案中,**不涉及**针对OCT4/SOX2或铁死亡的靶向策略。
3. **研究前沿**:探索铁死亡在卵巢癌治疗中的作用是活跃的研究领域。理论上,调控与肿瘤干细胞(可能涉及OCT4/SOX2等因子)相关的代谢和抗氧化通路,以增强其对铁死亡的敏感性,是一个有潜力的研究方向,但**尚处于临床前探索阶段**,距离临床转化甚远。
**总结**:将OCT4/SOX2与卵巢子宫内膜样癌的铁死亡直接关联,缺乏现有证据支持。临床医生应明确OCT4/SOX2在卵巢肿瘤诊断中的特定用途,并对子宫内膜样癌采取基于证据的规范治疗。关于铁死亡在该癌种中的应用,需关注未来高质量的基础与临床研究进展。
---
*本分析基于所提供的病理学与妇科病理学文献。关于OCT4/SOX2与铁死亡在卵巢癌中的潜在关联属于前沿研究假设,并非当前临床实践的一部分。*
生脉注射液
## 生脉注射液临床证据综合评估
生脉注射液是一种源自经典方剂“生脉散”的中成药注射剂,由红参、麦冬、五味子组成,具有益气养阴、复脉固脱的功效。以下基于《生脉类注射剂临床应用中国专家共识》及《中成药治疗社区获得性肺炎指南》等文献,对其临床应用证据进行结构化分析。
### 一、 核心药理学与物质基础
1. **主要成分**:红参、麦冬、五味子。
2. **关键活性成分**:经高效液相色谱法测定,其主要成分包括**人参皂苷Rg1、Re、Rb1**和**五味子醇甲**。
3. **作用机制研究**(临床前):
* **心肌保护**:抗缺血缺氧及再灌注损伤,抑制心肌细胞凋亡与纤维化[2]。
* **脑保护**:减轻脑缺血再灌注损伤,调节内质网应激与神经细胞凋亡[2]。
* **血管内皮保护**:人参皂苷是保护血管内皮缺氧复氧损伤的活性部位[2]。
* **抗休克**:抑制炎症反应,改善微循环,调节免疫功能[2]。
* **调节免疫与协同抗肿瘤**:可改善免疫功能,降低化疗药物引起的骨髓抑制等不良反应[2]。
### 二、 临床适应症与证据强度
以下证据主要来源于随机对照试验(RCT)及Meta分析。
| 适应症/应用场景 | 核心临床证据 | 推荐级别/备注 |
| :--- | :--- | :--- |
| **1. 心力衰竭** | • **慢性心衰**:多项RCT及Meta分析显示,联合常规西药可**进一步提高临床总有效率、增加LVEF、增加6分钟步行距离、降低NT-proBNP水平**,优于单用西药[2]。<br>• **急性心衰/难治性心衰**:RCT显示联合治疗可缩短症状缓解时间,改善血流动力学指标[2]。 | 专家共识推荐用于气阴两虚型心衰的辅助治疗。 |
| **2. 冠心病(心绞痛、心肌梗死)** | • **心绞痛**:Meta分析显示联合西医治疗可提高疗效,降低炎症指标[2]。<br>• **急性心肌梗死**:系统评价显示,联合常规治疗可**降低死亡率及心律失常、心源性休克、心力衰竭的发生率**[2]。 | 辅助治疗,改善预后。 |
| **3. 休克** | • **心源性休克**:RCT显示可增强心脏泵血功能,改善血液流变学,提高疗效[2]。<br>• **感染性休克**:小样本研究显示可改善氧代谢,调节炎症因子[2]。<br>• **低血容量性休克**:联合升压药可更持久稳定血流动力学[2]。 | 《CAP指南》推荐用于**严重CAP伴Yin depletion syndrome(面红、烦躁、热汗、低血压等)** (推荐强度:2C)[1]。 |
| **4. 肿瘤辅助治疗** | • **减轻化疗毒性**:Meta分析显示,联合化疗可**降低蒽环类药物心脏毒性**(改善心电图异常、LVEF),减轻恶心呕吐,改善白细胞降低[2]。<br>• **改善癌因性疲乏**:RCT显示有一定疗效[2]。 | 用于改善化疗相关不良反应及生活质量。 |
| **5. 缺血性中风** | • **急性期辅助治疗**:Meta分析显示,联合常规治疗可**提高临床总有效率、降低神经功能缺损评分、提高生活质量**[2]。 | 辅助治疗。 |
| **6. 病毒性心肌炎** | • Meta分析显示,联合西药治疗的**临床总有效率优于单用西药**,且症状及心肌酶改善更佳[2]。 | 辅助治疗。 |
| **7. 低血压** | • 对原发性低血压及血液透析中低血压,RCT显示可提高血压,减少透析低血压发生率[2]。 | 对症治疗。 |
| **8. 其他**(COPD、糖尿病并发症等) | • 小样本RCT提示对慢性阻塞性肺疾病急性加重期、2型糖尿病并发症等有改善作用[2]。<br>• 被列入多版《新型冠状病毒肺炎诊疗方案》重型/危重型“内闭外脱证”推荐用药[2]。 | 证据级别较低,需进一步研究。 |
### 三、 用法用量与安全性
1. **用法用量**:
* **静脉滴注**:一次**20-100 mL**,用**5%葡萄糖注射液250-500 mL**稀释后使用[1][3]。
* **滴速控制**:不宜超过**60滴/分钟**[1]。
* **药品说明书**:标明一次**25-60 mL**,用5%葡萄糖注射液250-500 mL稀释[3]。
2. **安全性**:
* **总体不良反应率**:大规模集中监测显示为**0.176%-1.23%**,属偶见水平[2]。
* **主要不良反应**:以**速发型过敏反应**为主,多在用药30分钟内出现。临床表现包括:
* 全身性损害(发热、寒战等)
* 皮肤及附件损害(皮疹、瘙痒)
* 心血管系统损害(心悸、血压波动)
* 消化系统损害、呼吸系统损害等[2]。
* **严重不良反应**:需警惕**过敏性休克**的可能。
* **禁忌**:
* 对本品过敏或严重不良反应病史者禁用。
* 药品说明书强调:**有实证及暑热等病热邪尚盛者、咳而尚有表证未解者禁用**[3]。
### 四、 结论与临床应用要点
1. **证据总结**:生脉注射液在**心力衰竭、冠心病、休克、肿瘤化疗辅助、缺血性中风**等多个领域,有相对较多的RCT和Meta分析证据支持其作为联合用药能改善临床症状、提高疗效、减少不良反应。
2. **中医辨证要点**:其核心适应症为**气阴两虚证**或**阴竭证**。在休克或严重感染中的应用,需严格对应中医辨证(如《CAP指南》中所述的Yin depletion syndrome)[1]。
3. **使用警示**:
* **超说明书用药**:部分临床应用(如肿瘤辅助、中风)属于**超说明书用药**,需充分评估获益风险,并遵循相关管理规定。
* **安全第一**:用药前应详细询问过敏史,**首次用药或更换批次时建议缓慢滴注并密切观察**。备好肾上腺素等抢救药品。
* **溶媒选择**:严格使用说明书推荐的**5%葡萄糖注射液**稀释,避免配伍禁忌。
---
**免责声明**:以上信息基于提供的文献资料合成,旨在为临床专业人员提供参考。生脉注射液的使用必须严格遵循药品说明书,并在中医辨证指导下进行。超说明书用药应具备充分的临床理由并符合医院相关规定。用药过程中需密切监测不良反应。
生脉注射液对脓毒性休克中血管内皮细胞铁死亡与线粒体自噬、代谢重编程的交互网络
## 生脉注射液对脓毒性休克中血管内皮细胞铁死亡、线粒体自噬及代谢重编程的潜在干预机制分析
基于现有文献,生脉注射液对脓毒性休克(感染性休克)的疗效已有临床观察支持,但其对血管内皮细胞铁死亡、线粒体自噬及代谢重编程这一复杂交互网络的具体作用机制,**尚未有直接、系统的研究证据**。以下分析是根据其已知的药理作用,结合脓毒性休克内皮损伤的病理生理学,进行的**理论推演和潜在机制假说**。
### 一、 生脉注射液治疗脓毒性休克的临床证据基础
1. **临床疗效**:多项小样本随机对照试验(RCT)表明,生脉注射液联合常规治疗可改善感染性休克患者的氧代谢、调节血清炎症因子(如TNF-α, IL-6)、降低内皮素-1(ET-1)水平、提高一氧化氮(NO)表达,从而提高临床总有效率[2]。
2. **作用定位**:中医理论认为其适用于“气阴两亏,脉虚欲脱”的证候,这与脓毒性休克后期出现的循环衰竭、组织灌注不足、能量代谢障碍有相通之处。
### 二、 对血管内皮细胞铁死亡的潜在干预
铁死亡的核心是**脂质过氧化与抗氧化防御失衡**。生脉注射液的成分可能从以下环节介入:
1. **增强抗氧化防御系统(对抗脂质过氧化)**:
* **理论依据**:生脉注射液的主成分**人参皂苷**(如Rb1, Rg1)和**五味子醇甲**在临床前研究中被证实具有明确的抗氧化作用,能够**降低丙二醛(MDA)含量、减少活性氧(ROS)生成**[2]。
* **潜在靶点**:
* **直接清除ROS**:其活性成分可能作为自由基清除剂。
* **上调内源性抗氧化酶**:可能通过激活**Nrf2/ARE通路**,上调**谷胱甘肽过氧化物酶4(GPX4)**、**血红素加氧酶-1(HO-1)** 等关键抗氧化蛋白的表达,从而增强细胞对脂质过氧化的抵抗能力。
* **提供还原当量**:通过改善细胞能量代谢(见下文),可能间接维持**NADPH**的供应,这对于GPX4和**铁死亡抑制蛋白1(FSP1)** 系统的功能至关重要。
2. **调节铁代谢(减少催化铁)**:
* **理论依据**:细胞内游离铁(Fe²⁺)是芬顿反应催化脂质过氧化的关键。生脉注射液改善微循环和细胞活力的作用[2],可能有助于维持正常的铁代谢稳态。
* **潜在靶点**:可能通过减轻炎症和氧化应激,间接稳定**铁蛋白**,抑制**核受体共激活因子4(NCOA4)** 介导的铁蛋白自噬,从而减少铁离子的异常释放。
### 三、 对线粒体自噬的潜在调节
线粒体自噬在脓毒症中是一把“双刃剑”。生脉注射液可能促进其保护性作用。
1. **改善线粒体功能,减少自噬负荷**:
* **理论依据**:注射用益气复脉(冻干)可**调节线粒体功能障碍和蛋白激酶Cδ/线粒体动力相关蛋白1(PKCδ/Drp1)介导的线粒体过度裂变**[2]。
* **潜在机制**:通过抑制Drp1介导的**过度线粒体分裂**,可能减少因结构严重破坏而需要被清除的线粒体数量,从而**避免过度或失调的线粒体自噬**,维持足够的线粒体数量以保障ATP生产。
2. **可能促进选择性清除严重受损线粒体**:
* **理论依据**:其成分能增加内皮细胞活力,减少缺氧复氧损伤[2]。
* **潜在机制**:通过改善整体细胞能量和氧化还原状态,可能有助于维持**PINK1-Parkin**等经典线粒体自噬通路的正常功能,使其更精确地识别和清除真正功能衰竭的线粒体,而非引发大规模清除。
### 四、 对代谢重编程的潜在纠正
脓毒症中内皮细胞向糖酵解增强的代谢重编程,削弱了抗氧化能力。
1. **改善能量代谢与线粒体呼吸**:
* **直接证据**:在脓毒症模型中,注射用益气复脉(冻干)可**抑制调控线粒体呼吸链复合物的异常,恢复ATP的产生**[2]。
* **意义**:这直接对抗了脓毒症导致的线粒体功能障碍和能量危机,可能有助于将细胞代谢从单纯的糖酵解向更高效的氧化磷酸化(OXPHOS)部分回拨。
2. **增加NADPH供应潜力**:
* **间接机制**:通过改善线粒体功能和整体能量状态,可能促进葡萄糖代谢流向**磷酸戊糖途径(PPP)**,从而增加**NADPH**的生成。NADPH是维持GPX4和FSP1系统功能的核心辅因子。
### 五、 整合作用机制假说图
生脉注射液可能通过多靶点作用于脓毒性休克内皮损伤网络:
```
脓毒性休克应激
↓
[生脉注射液干预]
├─ 成分(人参皂苷、五味子醇甲)
│
├─→ **抗氧化** (↓ROS, ↓MDA, ↑Nrf2/GPX4) → **抑制铁死亡执行**
│
├─→ **稳线粒体** (↓PKCδ/Drp1过度分裂) → **维持功能线粒体池** & **避免过度自噬**
│
└─→ **调代谢** (恢复呼吸链功能,↑ATP) → **改善能量供应** & **潜在↑NADPH**
↓
[内皮细胞结局改善]
• 脂质过氧化减轻
• 线粒体质量与功能改善
• 能量代谢部分恢复
• 细胞活力增加,屏障功能改善
```
### 六、 结论与展望
1. **机制推测性质**:上述分析是基于生脉注射液已知的抗氧化、线粒体保护、改善微循环等作用,与脓毒症内皮损伤的分子病理环节进行的**合理关联和假设**,**并非已有确凿的直接实验证据**。
2. **整体调节优势**:生脉注射液的特点可能在于其**多成分、多靶点**的整体调节作用,恰好对应了脓毒性休克内皮损伤中**铁死亡、自噬、代谢重编程三者交织的复杂网络**,而非针对单一通路。
3. **临床意义与研究方向**:
* **临床意义**:现有临床研究支持其在感染性休克中的辅助治疗价值,其潜在机制可能部分通过保护内皮、改善微循环实现。
* **研究缺口**:亟需设计严谨的临床前研究,在脓毒症内皮细胞和动物模型中,直接验证生脉注射液对**GPX4活性、脂质过氧化水平、线粒体自噬流、细胞铁含量及代谢谱**的具体影响,以证实或修正上述假说。
* **治疗定位**:在临床实践中,它应作为**综合抗休克治疗(液体复苏、抗感染、血管活性药物等)的辅助**,用于符合气阴两虚证候的患者,而非替代核心治疗。
---
**免责声明**:本分析是基于现有药理学研究和病理生理学理论进行的机制性推测,旨在为后续基础研究提供思路。生脉注射液在脓毒性休克中的应用必须严格遵循临床指南和药品说明书,并在中医辨证指导下进行。其对抗内皮细胞铁死亡等具体分子机制尚未经实验证实,不能作为临床决策的直接依据。