结直肠癌MMR蛋白缺失分析
结直肠癌患者,病理报告显示MMR蛋白(MLH1, PMS2, MSH2, MSH6)中MLH1和PMS2缺失。请问这提示什么?下一步应该建议患者做什么检测来排除Lynch综合征?
根据检索到的临床指南与共识,针对您描述的结直肠癌患者病理结果(MLH1与PMS2蛋白表达缺失),其临床意义及后续诊断流程分析如下。
## 临床意义解读
免疫组化(IHC)检测显示 **MLH1和PMS2蛋白同时缺失**,是结直肠癌中最常见的错配修复缺陷(dMMR)表达模式之一[8]。这种模式高度提示肿瘤为**微卫星高度不稳定(MSI-H)型**[1][9]。
其临床意义主要包括:
1. **预后与治疗指导**:dMMR/MSI-H的II期结直肠癌患者通常预后较好[8]。更重要的是,dMMR状态是预测患者能否从免疫检查点抑制剂治疗中获益的关键生物标志物[8][10]。
2. **遗传风险评估**:MLH1/PMS2缺失可能由两种主要原因导致:
* **散发型(体细胞改变)**:约占大多数,主要由**MLH1基因启动子区异常甲基化**引起[2][10]。
* **遗传型(胚系突变)**:即**林奇综合征(Lynch Syndrome, LS)**,由MLH1等基因的胚系致病突变导致[5][7]。
因此,发现MLH1/PMS2缺失后,核心任务是**鉴别该dMMR状态是散发性的还是遗传性的(即林奇综合征)**,这对患者及其家属的癌症风险管理至关重要。
## 下一步诊断流程与建议
基于多部国际与国内指南共识,推荐遵循以下标准化流程进行鉴别诊断[2][6][7][12]:
**以下流程图概述了基于肿瘤MMR蛋白IHC和MSI结果的林奇综合征诊断路径:**
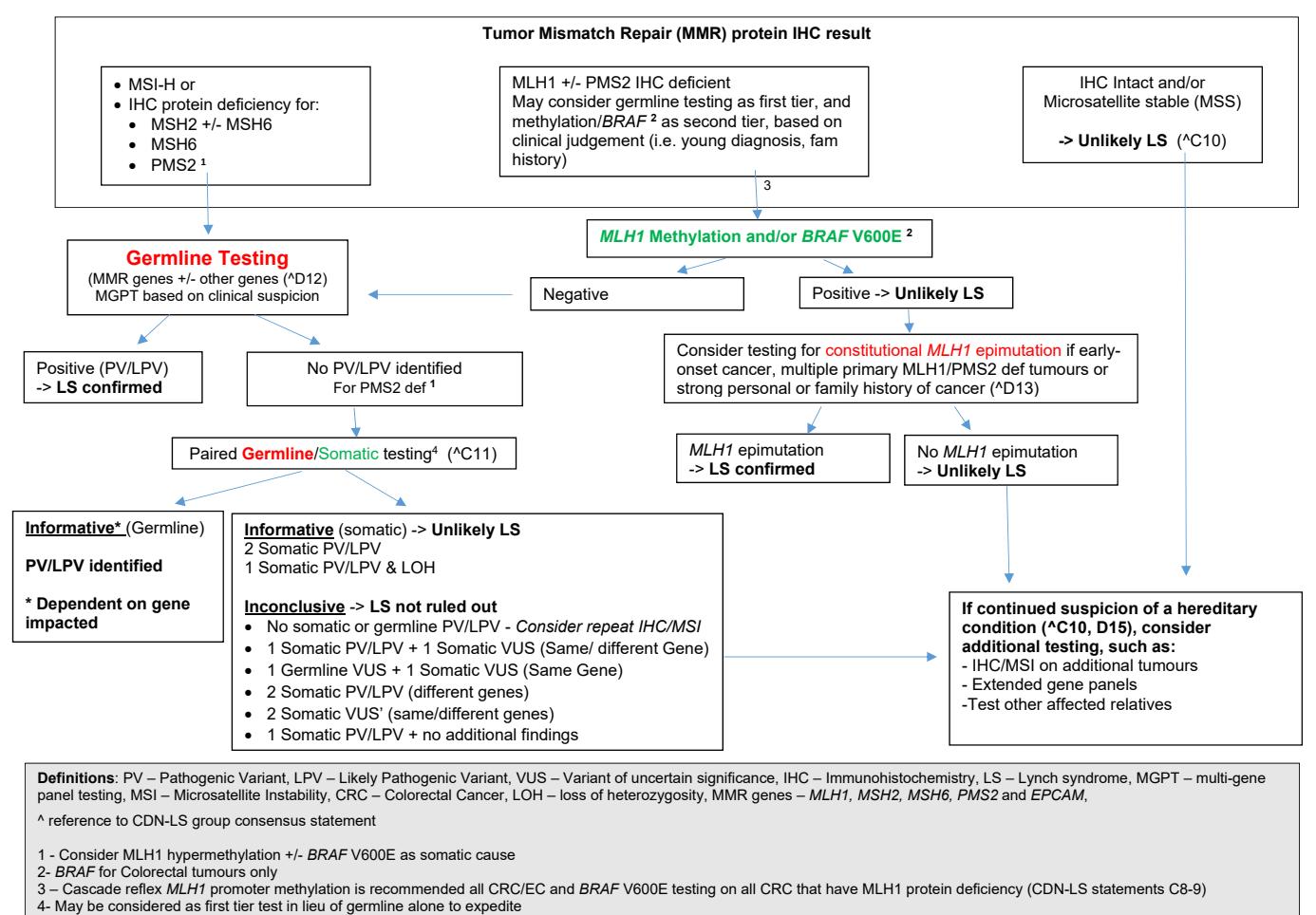
*Figure: 基于肿瘤错配修复蛋白免疫组化和微卫星不稳定性结果的林奇综合征临床诊断流程图*
### 步骤一:进行MLH1启动子甲基化或BRAF V600E突变检测
这是区分散发性和遗传性病例的关键一步。
* **检测选择**:对MLH1/PMS2缺失的结直肠癌,应进行 **MLH1启动子甲基化检测** 或 **BRAF V600E突变检测**[2][6][10]。
* **结果解读与后续行动**:
* **如果检测阳性**(即存在MLH1启动子高甲基化或BRAF V600E突变):这**强烈支持散发型结直肠癌**,林奇综合征的可能性极低[2][10][14]。通常无需进行胚系基因检测。
* **如果检测阴性**(即MLH1启动子未甲基化且BRAF为野生型):则**不能排除林奇综合征**,需进入下一步[6][12]。
### 步骤二:进行林奇综合征相关基因的胚系突变检测
当上述检测为阴性时,应建议患者进行遗传咨询并接受**胚系基因检测**,这是诊断林奇综合征的金标准[7][12]。
* **检测基因**:至少应包括 **MLH1、MSH2、MSH6、PMS2** 及 **EPCAM** 基因[4][5][13]。
* **检测意义**:若在血液样本中检测到上述任一基因的胚系致病突变,即可确诊为林奇综合征[5]。这要求对患者及其高危亲属启动终身癌症监测计划。
## 关键生物学机制背景
MMR蛋白以异源二聚体形式发挥作用。MLH1与PMS2形成MutLα复合物,MSH2与MSH6形成MutSα复合物[3]。MLH1是复合物的稳定核心,因此MLH1的失活(无论是通过甲基化还是突变)通常会导致其伙伴蛋白PMS2的继发性降解,在IHC上表现为两者共缺失[8]。
**以下示意图展示了DNA错配修复系统中MutLα和MutSα复合物的蛋白亚基组成:**

*Figure: DNA错配修复通路中形成MutLα和MutSα复合物的蛋白亚基示意图*
## 病理示例参考
**以下病理图像展示了结直肠癌组织中MMR蛋白表达的典型模式,特别是PMS2的单独缺失:**
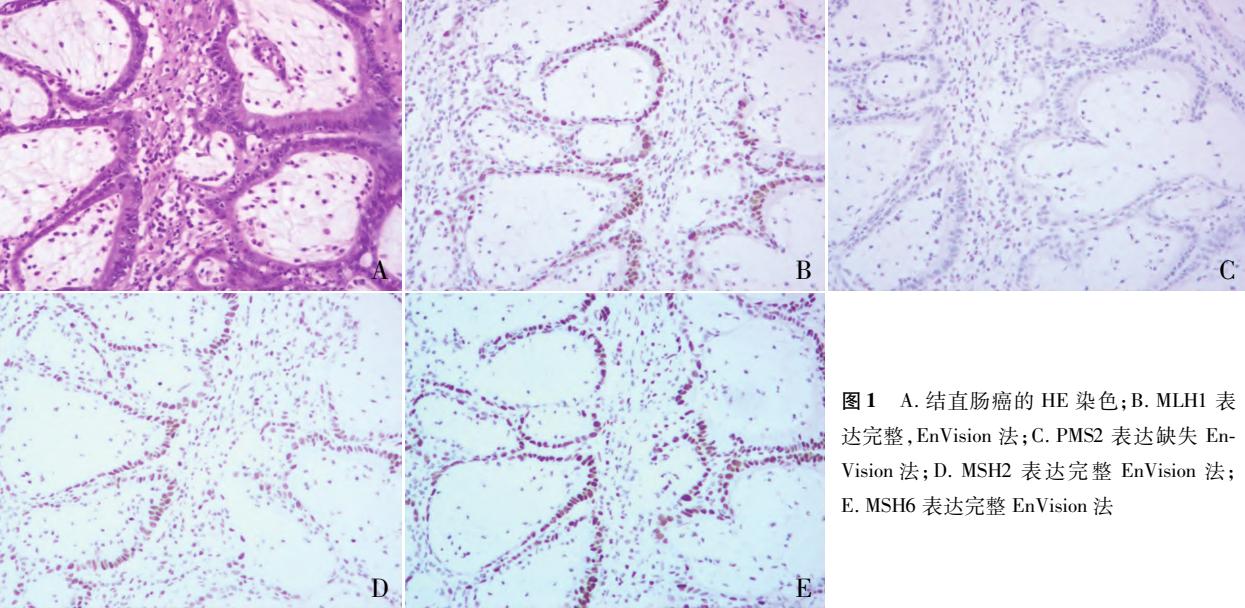
*Figure: 结直肠癌组织病理学特征及MMR蛋白表达分析,结果显示PMS2蛋白表达缺失*
## 总结与建议
1. **初步判断**:结直肠癌IHC显示MLH1与PMS2共缺失,提示dMMR/MSI-H状态,需启动林奇综合征鉴别诊断流程。
2. **首选检测**:建议对该肿瘤组织进行 **MLH1启动子甲基化检测** 或 **BRAF V600E突变检测**。
3. **后续行动**:
* 若甲基化/BRAF检测为阳性,倾向于散发癌,可基本排除LS。
* 若检测为阴性,应**强烈建议患者接受遗传咨询并进行MLH1等基因的胚系检测**以明确诊断。
4. **临床管理**:无论最终是否为LS,dMMR状态本身已为患者的预后判断和免疫治疗选择提供了关键信息。
---
**进一步诊断建议(仅供参考)**
为完成上述鉴别诊断流程,需要获取以下关键信息:
* **肿瘤组织的MLH1启动子甲基化状态或BRAF V600E突变状态**:这是决定是否需要启动胚系基因检测的分水岭。
* **详细的个人及家族肿瘤史**:包括患者本人其他原发性肿瘤史,以及一级、二级亲属中结直肠癌、子宫内膜癌、胃癌、卵巢癌等LS相关肿瘤的发病情况与年龄,用于综合风险评估。
---
*注:以上分析基于检索到的临床指南与文献,旨在为临床决策提供参考。所有诊断与检测建议均需在临床医生结合患者具体情况进行全面评估后实施。*
MLH1/PMS2蛋白缺失但MLH1启动子未甲基化患者中,最终确诊为林奇综合征的比例及相关的临床病理特征
根据检索到的文献,针对结直肠癌或子宫内膜癌中MLH1/PMS2蛋白缺失但MLH1启动子未甲基化患者的确诊比例及临床特征,分析如下。
## 确诊林奇综合征的比例
在免疫组化(IHC)显示MLH1/PMS2蛋白缺失的肿瘤患者中,进行MLH1启动子甲基化检测是鉴别散发性和遗传性病例的关键步骤。
* **总体情况**:一项纳入1,336例子宫内膜癌的研究显示,在380例错配修复缺陷(MMRd)的患者中,275例(72.4%)为MLH1高甲基化的散发型病例,而**仅有36例(9.5%)最终确诊为林奇综合征**[3]。这提示,在IHC提示MLH1/PMS2缺失的子宫内膜癌患者中,绝大多数为散发型。
* **在未甲基化亚组中的比例**:当MLH1启动子甲基化检测结果为阴性(即未甲基化)时,患者为林奇综合征的可能性显著增加。检索到的共识明确指出,对于IHC显示MLH1蛋白表达缺失的患者,应进行MLH1启动子甲基化检测以评估是否为散发型[3]。若结果为阴性,则不能排除林奇综合征,需进一步进行胚系基因检测[1][3]。虽然未提供该亚组(MLH1/PMS2缺失且未甲基化)中林奇综合征的确切阳性预测值,但临床路径将其视为需要启动胚系检测的高危人群。
**以下流程图清晰地展示了基于肿瘤MMR蛋白IHC和MSI结果的林奇综合征标准化诊断路径,其中MLH1/PMS2缺失且甲基化/BRAF检测阴性是启动胚系检测的关键节点:**

*Figure: 基于肿瘤错配修复蛋白免疫组化和微卫星不稳定性结果的林奇综合征临床诊断流程图*
## 相关的临床病理特征
与散发型子宫内膜癌相比,林奇综合征相关子宫内膜癌(LS-EC)具有以下特征:
1. **发病年龄更早**:LS-EC患者的平均诊断年龄为**45-55岁**,显著早于散发型患者。一项研究显示,LS-EC的诊断平均年龄为48.5岁,比散发型提早约10年[3]。
2. **缺乏典型高危因素**:LS-EC患者常**无肥胖、高血压、糖尿病**等与雌激素过度刺激相关的经典高危因素[3]。
3. **肿瘤部位倾向**:LS-EC更倾向于发生在**子宫下段**。一项研究发现,在子宫下段子宫内膜癌患者中,MSH2突变携带者(林奇综合征的一种类型)的比例(14.2%)远高于非林奇综合征患者(1.8%)[3]。
4. **组织学类型**:尽管以**子宫内膜样癌**最为常见(约占89.2%),但也可发生非子宫内膜样癌,如透明细胞癌、浆液性癌等[3]。
5. **基因特异性风险**:不同MMR基因突变导致的癌症风险不同。携带**MSH2或MSH6胚系突变**的女性患子宫内膜癌的终生累积风险较高,而MLH1和PMS2突变携带者的风险相对较低[1][2]。具体累积风险数据如下表所示:
| MMR基因 | 75岁前患子宫内膜癌的累积风险(女性) | 结直肠癌终生风险(女性/男性) |
| :--- | :--- | :--- |
| **MLH1** | 37.0%[3] | 48.3% / 57.1%[2] |
| **MSH2/EPCAM** | 48.9%[3] | 46.6% / 51.4%[2] |
| **MSH6** | 41.1%[3] | 20.3% / 11.7%[2] |
| **PMS2** | 12.8%[3] | 10.4% / 数据缺失[2] |
## 总结与临床建议
1. **确诊比例**:在IHC显示MLH1/PMS2缺失的肿瘤中,大部分(约70%)为MLH1启动子甲基化阳性的散发型。在**甲基化阴性的患者中,林奇综合征的可能性显著增高**,是胚系基因检测的明确指征。
2. **高危特征**:对于MLH1/PMS2缺失且甲基化阴性的患者,若同时具备**发病年龄轻(<50岁)、肿瘤位于子宫下段、缺乏典型内分泌高危因素**等临床特征,应高度警惕林奇综合征。
3. **标准流程**:临床处理应遵循标准化路径(如上图所示):IHC初筛发现MLH1/PMS2缺失 → 进行MLH1启动子甲基化检测 → 若结果为阴性 → **建议遗传咨询并接受MMR基因(MLH1, MSH2, MSH6, PMS2及EPCAM)的胚系检测**以明确诊断[1][3]。
---
*注:以上分析基于检索到的专家共识,主要数据来源于子宫内膜癌人群。结直肠癌中林奇综合征的比例和特征可能有所不同,但鉴别诊断的分子逻辑(甲基化检测)是相似的。所有诊断决策需结合患者完整的个人史和家族史,并在遗传咨询后进行。*