早产儿女,小于胎龄儿,双胎之小,查体双手通贯掌,头围大,前囟宽,鼻梁低,双手通贯掌,主动脉缩窄,考虑
早产儿女,小于胎龄儿,双胎之小,查体双手通贯掌,头围大,前囟宽,鼻梁低,双手通贯掌,主动脉缩窄,考虑什么
根据您提供的临床信息,该患儿(早产、小于胎龄儿、双胎之小)合并多种先天性异常,包括**双手通贯掌、头围大、前囟宽、鼻梁低、主动脉缩窄**。这些特征高度提示存在一种**染色体异常或遗传综合征**。
### 鉴别诊断分析
结合提供的临床特征与现有文献证据,主要考虑以下诊断:
1. **唐氏综合征 (Down Syndrome, Trisomy 21)**
* **核心特征匹配**:您描述的“双手通贯掌”是唐氏综合征的典型皮肤纹理特征之一[6]。其他特征如“鼻梁低平”也完全符合其特殊面容的描述[6][10]。主动脉缩窄虽非其最常见的心脏畸形(室间隔缺损、房室间隔缺损更常见),但唐氏综合征患者中先天性心脏病的总体发生率约为50%[10],且可涵盖多种类型。
* **支持证据**:文献明确指出唐氏综合征的临床表现包括“特殊面容,如面部扁平...鼻梁低平”以及“特殊的皮肤纹理,如贯通手”[6]。此外,约30%的患者伴有先天性心脏病[6]。
* **需完善检查**:确诊需进行染色体核型分析,标准型为47,XX(XY),+21[6]。
2. **特纳综合征 (Turner Syndrome, 45,XO)**
* **核心特征匹配**:“主动脉缩窄”是特纳综合征最具特征性的心血管畸形之一[5][7][11]。“头围大”可能需鉴别,但特纳综合征更典型的特征是身材矮小、颈蹼、后发际低等。
* **支持证据**:特纳综合征的主要症状包括“先天性心脏缺陷(主动脉缩窄、二尖瓣或主动脉瓣病变)”以及“特殊的躯体特征”[11]。然而,典型特纳综合征患者为女性,且“双手通贯掌”并非其标志性表现。
* **需完善检查**:确诊需进行染色体核型分析,典型核型为45,XO[6]。对于有主动脉缩窄的女性患儿,特纳综合征是必须排除的诊断[7]。
3. **22q11.2缺失综合征 (DiGeorge综合征)**
* **核心特征匹配**:“主动脉缩窄”可作为其心脏圆锥干畸形的一种表现[2][5]。该综合征常伴有特殊面容(如眼距宽、鼻梁低平)、胸腺发育不良导致的免疫缺陷、低钙血症等。
* **支持证据**:22q11.2缺失综合征的异常表现包括“心脏圆锥干畸形、腭裂”以及“面中部发育不全”[2]。其心血管畸形中,主动脉弓异常(包括缩窄)是已知并发症[5]。
* **需完善检查**:需进行染色体微阵列分析(CMA)或FISH检测22q11.2区域缺失。
4. **努南综合征 (Noonan Syndrome)**
* **核心特征匹配**:此为常染色体显性遗传的RASopathy,常见特征包括特殊面容(眼距宽、眼睑下垂、低耳位)、身材矮小、先天性心脏病(以肺动脉瓣狭窄最常见,但也可有其他类型)[1][5][9]。
* **支持证据**:努南综合征患者中,心脏疾病关联率高达80%,其中60%为先天性心脏病[5]。虽然典型面容与唐氏综合征不同,但部分特征(如鼻梁低)可能重叠。
* **需完善检查**:需进行RAS-MAPK通路基因(如PTPN11, SOS1, RAF1等)的分子遗传学检测[1][9]。
### 诊断思路建议
1. **首要检查**:**染色体核型分析**。这是鉴别唐氏综合征、特纳综合征及其他染色体数目异常的金标准[6][11]。鉴于“通贯掌”这一强提示,应优先排查唐氏综合征。
2. **若染色体核型正常**,则需考虑:
* **微缺失/微重复综合征**:进行**染色体微阵列分析(CMA)**,以检测22q11.2缺失等拷贝数变异。
* **单基因遗传病**:根据进一步表型(如生长发育、智力、其他畸形),考虑进行**目标基因Panel测序**(如努南综合征多基因Panel[9])。
3. **专科评估**:
* **心脏超声**:明确主动脉缩窄的详细解剖及其它可能并存的心内畸形。
* **遗传咨询**:对患儿父母进行遗传咨询,解释再发风险,并讨论可能的产前诊断方案。
### 结论
该患儿的临床表现组合(通贯掌、特殊面容、先天性心脏病、小于胎龄儿)强烈指向**染色体异常或遗传综合征**。**唐氏综合征**因“通贯掌”这一高度特异性体征成为首要怀疑对象,但必须通过**染色体核型分析**与特纳综合征、22q11.2缺失综合征等其他疾病进行鉴别。
---
*本分析基于提供的文献资料生成,旨在为临床决策提供参考。所有诊断均需通过规范的遗传学检测确认,具体的诊疗方案请结合患儿全面情况由主管医生制定。*
digeorge综合征
基于提供的文献资料,以下是关于**DiGeorge综合征**的临床证据综述。
## 疾病定义与核心特征
DiGeorge综合征(DGS)是一种由胚胎期第3、4咽囊发育异常导致的多系统先天性疾病。其经典三联征包括:
1. **胸腺发育不全/缺失**:导致T细胞缺陷和免疫缺陷。
2. **甲状旁腺功能减退**:导致低钙血症。
3. **先天性心脏病**:主要为心脏圆锥干畸形[2]。
然而,临床表型具有高度变异性,并非所有患者均完全具备上述特征[2]。
## 病因与遗传学
DGS可由多种遗传和环境因素引起,构成一个“表型谱”。
**主要遗传病因:**
* **22q11.2缺失综合征(22q11.2DS)**:这是最常见的原因,约占DGS病例的90%[6]。其为常染色体显性遗传,多数(90-95%)为新发突变[2]。估计发病率约为1/4000活产儿[2]。
* **其他遗传综合征**:
* **CHARGE综合征**(常与*CHD7*基因突变相关)[1][2]。
* **10p部分单体综合征**[1][2]。
* **TBX1、TBX2、FOXI3、PAX1**等基因的单倍剂量不足或突变[2]。
**环境因素**:包括母体糖尿病胚胎病、宫内酒精或维甲酸暴露过量/不足等[2]。
以下示意图展示了22q11.2缺失综合征的基因组结构,有助于理解其分子遗传学基础:
**以下示意图展示了22q11.2缺失综合征的基因组结构:**
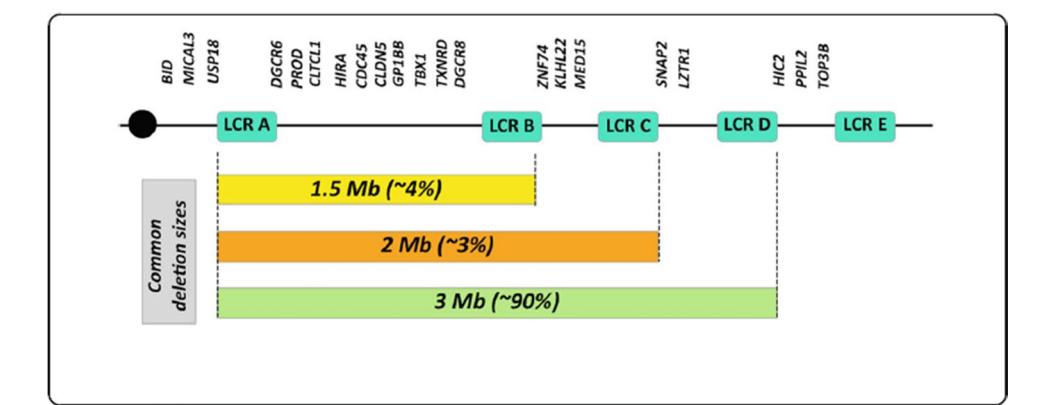
*Caption: 该图展示了22q11.2染色体区域的基因分布及三种常见微缺失的大小与发生频率。*
如图所示,90%的病例为LCR A至LCR D之间的3 Mb典型缺失,*TBX1*基因的缺失被认为是导致心脏畸形等核心表型的关键因素[图1]。
## 临床表现(扩展表型谱)
除经典三联征外,DGS/22q11.2DS可累及多个系统:
| 系统 | 常见异常表现 | 证据来源 |
| :--- | :--- | :--- |
| **心血管** | 心脏圆锥干畸形(如法洛四联症、永存动脉干、主动脉弓中断)、主动脉缩窄 | [1][2][3] |
| **免疫** | T细胞淋巴细胞减少(程度不一)、胸腺发育不全/缺失、免疫缺陷、易感染 | [2][6] |
| **内分泌** | 甲状旁腺功能减退(低钙血症)、甲状腺功能异常、生长激素缺乏 | [1][2] |
| **颅面与五官** | 腭裂/腭咽闭合不全、特殊面容(眼距宽、鼻梁低平、耳位低等)、后鼻孔闭锁(CHARGE)、听力损失、视力问题 | [1][2][4] |
| **发育与神经精神** | 发育迟缓、智力障碍(程度不一)、学习困难、自闭症谱系特征、精神疾病风险增高(如焦虑、精神分裂症) | [1][4][5] |
| **其他** | 喂养困难、生长迟缓、肾脏异常、骨骼畸形、血小板减少、自身免疫性疾病风险 | [1][2][5] |
## 诊断与评估
1. **临床诊断**:基于典型的多系统临床表现。
2. **遗传学确诊**:
* **一线检测**:**染色体微阵列分析(CMA)** 用于检测22q11.2缺失及其他拷贝数变异[4][5]。
* **其他方法**:荧光原位杂交(FISH)、多重连接依赖式探针扩增(MLPA)也可用于检测特定缺失[5]。
* **基因测序**:当表型典型但CMA阴性时,应考虑对*TBX1*、*CHD7*等相关基因进行测序[2]。
3. **多系统评估**:确诊后需进行全面的基线评估,包括心脏超声、免疫学功能(T/B/NK细胞计数、免疫球蛋白水平)、血钙/甲状旁腺激素、肾脏超声、发育评估等[4][5][6]。
## 免疫学分类与管理(关键更新)
最新的免疫管理指南建议摒弃“部分/完全/非典型DiGeorge”的旧术语,转而根据**T细胞淋巴细胞减少(TCL)的程度**进行描述性分类[6]:
* **正常T细胞数量**
* **轻度或显著的TCL**
* **严重的TCL(提示先天性无胸腺)**
**免疫管理核心建议:**
* **活疫苗接种**:仅适用于**无显著TCL**(即T细胞数量与功能接近正常)的患儿,并需在免疫专科医生评估后进行[6]。
* **先天性无胸腺症的治疗**:对于确诊为先天性无胸腺(如完全DiGeorge综合征)的患儿,**异基因加工胸腺组织移植(Rethymic®)** 可被视为一种医学必要的治疗选择[7]。
## 长期管理与随访
DGS/22q11.2DS需要终身、多学科的综合管理。成人患者指南强调[5]:
* **定期评估**:包括心血管、内分泌、免疫、精神神经、肾脏等多系统监测。
* **关注迟发性问题**:如主动脉根部扩张、帕金森病、精神分裂症、自身免疫病等。
* **功能支持**:评估认知、适应能力和日常功能,提供必要的教育、职业及社会心理支持。患者常存在“隐性残疾”,看似言语能力尚可,但执行功能、社交认知存在显著困难[5]。
* **围手术期注意**:因可能存在低钙血症和血小板异常,术前术后需监测血钙和全血细胞计数[5]。
## 遗传咨询
* **再发风险**:多数22q11.2DS为新发突变,但约5-10%为父母遗传所致[2]。因此,**必须对父母进行遗传学检测**以提供准确的再发风险评估[4][5]。
* **产前诊断**:对于有家族史的夫妇,可通过绒毛膜取样或羊膜腔穿刺进行产前遗传学诊断。
---
*本信息基于现有临床指南与文献合成,旨在支持临床决策。所有诊断与治疗决策需结合患者具体情况,由多学科团队制定。*
DiGeorge综合征患儿中,双手通贯掌等骨骼异常的发生率及临床意义
根据提供的文献资料,**关于DiGeorge综合征(22q11.2缺失综合征)患儿中“双手通贯掌”这一特定骨骼异常的发生率,现有检索到的证据中未提供明确的流行病学数据。**
### 现有证据分析
1. **核心表型与骨骼异常**:
* 文献明确列出了DiGeorge综合征相关的多种异常表现,包括**胸腺发育不全、心脏圆锥干畸形、腭裂、面中部发育不全、身材矮小、发育迟缓、血小板减少、认知及精神障碍**等[1]。
* 在提及的骨骼系统异常中,包括了**长骨增厚、骨髓腔变薄**(见于Kenny-Caffey综合征,一种与DiGeorge表型重叠的疾病)以及**薄骨干、三叶形颅骨**等[1]。
* **“双手通贯掌”作为一种特定的皮肤纹理(皮纹学)异常,并未在提供的DiGeorge综合征相关文献中被列为典型或常见特征**[1][2][3][4][5][6]。
2. **鉴别诊断的重要性**:
* “双手通贯掌”是**唐氏综合征(21-三体综合征)** 的经典且高特异性的体征之一[6]。在临床遇到具有此体征的患儿时,**首要的鉴别诊断应指向唐氏综合征**。
* 虽然DiGeorge综合征患儿可能因结缔组织异常而出现其他类型的骨骼或肢体异常(如指/趾畸形),但通贯掌并非其标志性表现。
### 临床意义与建议
1. **诊断思路**:对于一名疑似DiGeorge综合征(例如,有主动脉缩窄、低钙血症、特殊面容、免疫缺陷)但同时存在“双手通贯掌”的患儿,临床医生应考虑以下可能性:
* **表型重叠**:极少数情况下,可能存在更复杂的染色体异常或综合征,同时涵盖了两种疾病的部分特征。
* **偶然并存**:“通贯掌”在普通人群中也有一定比例(约1-2%),可能与DiGeorge综合征无关。
* **诊断再评估**:应重新审视核心诊断。**必须优先通过染色体核型分析排除唐氏综合征**,因为通贯掌对该病的提示价值更高。
2. **管理建议**:
* **完善遗传学检测**:不应仅凭“通贯掌”否定或支持DiGeorge综合征的诊断。确诊依赖于**染色体微阵列分析(CMA)** 以检测22q11.2缺失[4][5]。
* **全面评估**:无论是否存在通贯掌,对疑似DiGeorge综合征的患儿都应进行标准的多系统评估(心脏、免疫、内分泌、肾脏等)[4][5][6]。
* **遗传咨询**:需向家长解释,单一的非典型体征(如通贯掌)不改变DiGeorge综合征的核心管理原则,但可能提示需要进行更广泛的遗传学分析以排除其他综合征。
### 结论
当前证据表明,**“双手通贯掌”并非DiGeorge综合征(22q11.2缺失综合征)的典型或常见特征**,其在该病人群中的具体发生率未知。当此体征出现时,**其首要临床意义在于提示需重点鉴别唐氏综合征**。DiGeorge综合征的诊断应基于其核心表型(心脏畸形、免疫缺陷、低钙血症等)并通过CMA检测确认,而非依赖此皮纹学特征。
---
*本分析基于现有文献,旨在为临床鉴别诊断提供参考。具体的诊断应结合患儿全部临床表现和遗传学检测结果综合判断。*